Amplicon Sequencing (16S, 18S, ITS)
Amplicon sequencing is frequently used to identify and differentiate microbial species. Short (< 500 bp) hypervariable regions of conserved genes or intergenic regions are amplified by PCR, analyzed using NGS technology, and the resulting sequences are compared against microbial databases.
For bacteria and archaea, the 16S rRNA gene is the most common target for amplicon sequencing. For fungi, three targets are generally used: the 18S rRNA gene, and two internal transcribed spacers (ITS) located between rRNA genes. These regions are usually sufficiently divergent to separate even highly related species, and can sometimes differentiate subspecies.
Sequencing using MiSEQ
Paired End 300 base Read length
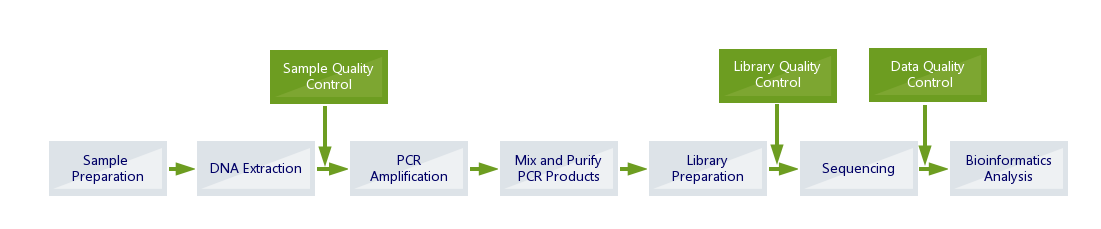
Standard Bioinformatics Included
Amplicon Metagenome Targets
Standard Metagenome Amplicon Bioinformatics:
Quality Control
- Raw data QC.
- Removal of low quality reads and trimming of Low quality bases.
- Adaptor trimming.
Metagenome Amplicon Analysis:
- Read 1 and 2 merging and clustering, removal of duplicates, chimera removal for Deep datasets of 16S (For bacterial), 18S (Archeal), ITS (Fungal) sequencing of the samples for complete.
- Taxonomic classification & Phylogenetic analysis.
- Diversity & Abundance estimation data analysis should be done using optimized and published pipelines.
- ALPHA & BETA Diversity analysis, 2d & 3D PCA analysis, HEATMAPS, Phylogenetic heat maps, statistical tests, hierarchal clustering etc.
- Functional prediction using 16S data (Picrust pipeline).
- Predicted genes will be tabulated and classified into functional categories from lower orders (individual genes) to higher orders (cellular processes).
- Relative abundance for each gene will be calculated for each individual gene. HEAT MAPS and statistical comparisons in groups.